
认识脊髓性肌萎缩症 科学规范全程防治
脊髓性肌萎缩症(SMA)是一种常染色体隐性遗传的神经肌肉疾病,发病率为1/6000~1/10000,人群致病基因携带率高达1/40~1/50。这意味着每40~50人中即有一名携带者,且携带者一般无临床症状、不易被发现。
发病机制
人体肢体活动依赖肌肉收缩完成,脊髓运动神经元是调控肌肉运动的核心中枢。正常人体内的“运动神经元存活1号”的基因(SMN1)能够制造出“运动神经元存活”(SMN)的蛋白,维持运动神经元存活与功能稳定,保障肌肉正常运动。
SMA患者因SMN1基因缺失或变异,导致体内SMN蛋白缺失或显著减少。运动神经元持续退化、萎缩甚至凋亡,肌肉失去神经支配后逐渐无力、萎缩,最终引发各种运动功能障碍的症状。
SMN1有个孪生姐妹—SMN2,它们的基因序列几乎完全一样,均可表达SMN蛋白,但SMN2基因仅能合成约10%是稳定的正常全长度SMN蛋白。当SMN1基因缺失时,机体只能依靠SMN2基因表达的蛋白。当合成的蛋白无法满足生理需求,就会引发疾病。SMN2基因拷贝数对确诊SMA新生儿的后续临床治疗有重要指导价值,因此筛查阳性新生儿的确诊试验应包含SMN2基因拷贝数信息。
症状及分型
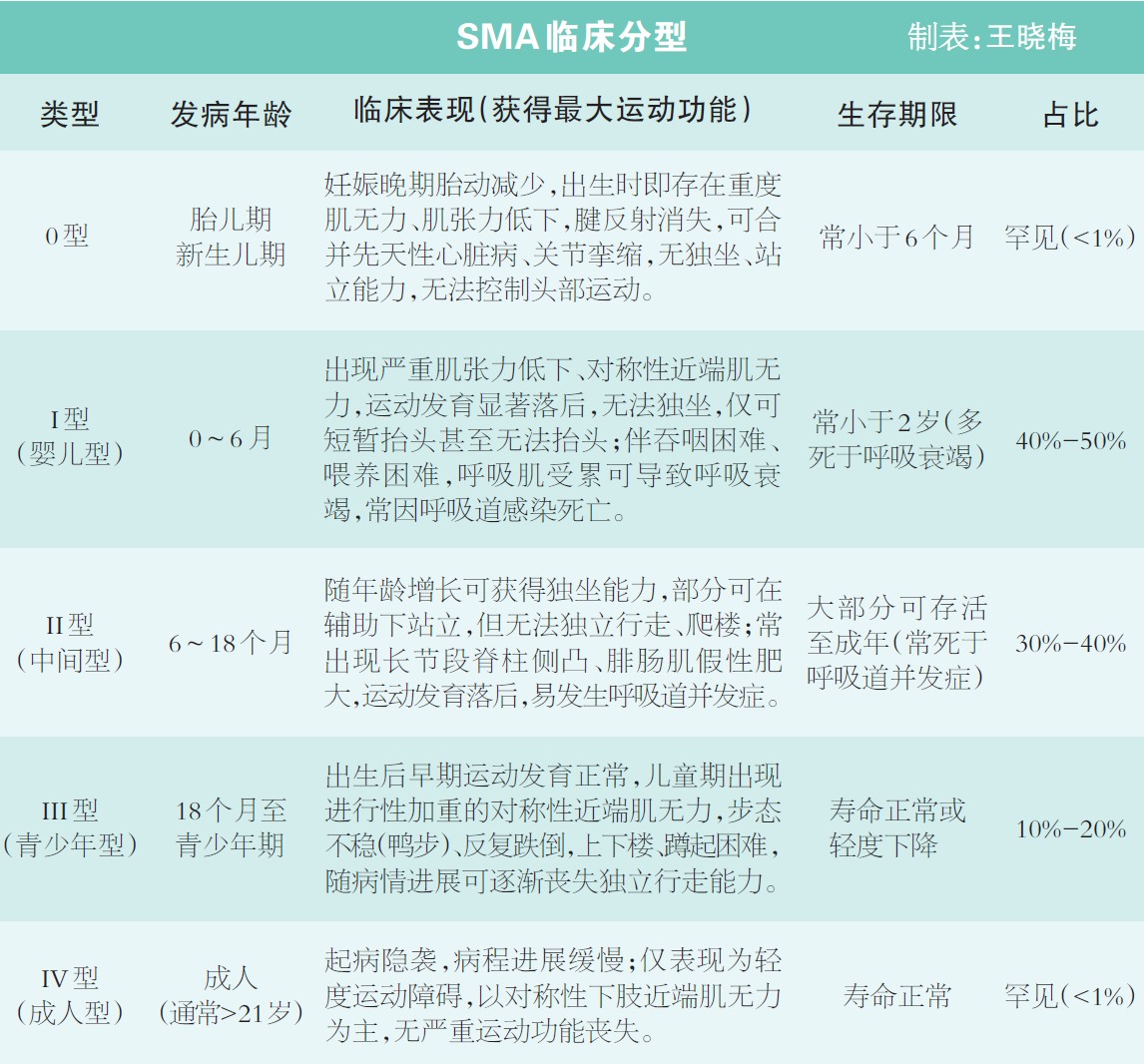
SMA的典型表现是四肢无力、肌肉萎缩,站立不能、行走受限等,因呼吸肌无力常合并肺部感染,因咀嚼肌无力常伴有营养障碍,可能同时存在生长发育迟缓、脊柱侧弯、关节挛缩、髋关节半脱位等情况。结合临床典型表现,根据发病年龄和运动功能里程碑分为5型,其中0型患者通常于新生儿期死亡,Ⅰ型SMA是最常见的类型,Ⅳ型则非常罕见,各型具体特征存在明显差异(见下表)。
治疗现状
随着医学发展,SMA已从“无药可治”转变为可防可治的遗传性疾病,目前临床上拥有药物、康复、呼吸及营养支持的综合诊疗体系。
药物治疗主要包括三类:一是诺西那生钠,通过鞘内注射修正异常的RNA剪接过程,让SMN2基因生产更多的SMN蛋白,改善患者运动功能;二是利司扑兰,为口服小分子药物,可系统性提升全身SMN蛋白水平,给药便捷、患者依从性更高;三是基因治疗药物(如Zolgensma),通过一次性导入正常基因,从根本上解决基因缺陷问题。适用于特定类型的SMA患者,尤其是年龄较小的患儿,有望实现长期甚至一次性的治疗效果。
康复干预是长期管理的关键。规律开展肌肉牵伸、运动训练,可预防肌肉挛缩与关节畸形;矫形器具可辅助维持正确的姿势与运动功能;咳嗽和排痰训练能改善呼吸功能,减少呼吸道感染的风险。同时,需长期开展呼吸功能监测与通气支持,针对吞咽困难患者制定个性化营养方案,必要时采用鼻管饲、胃造瘘等方式保障营养供给。
预防措施
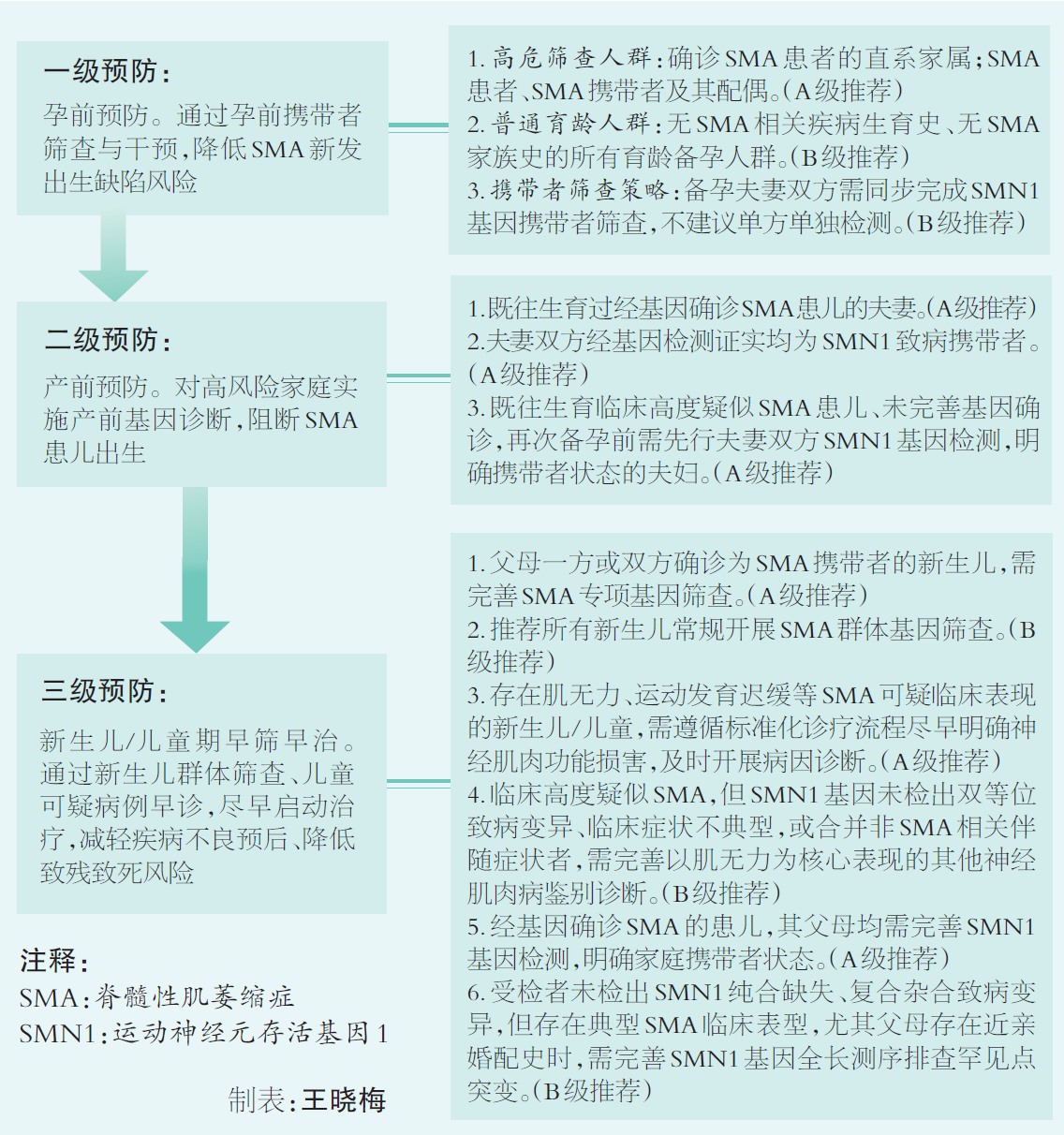
依据2023版《脊髓性肌萎缩症中国三级预防指南》,我国实行三级预防体系:一级预防针对高危人群和一般人群进行扩展性携带者筛查(ECS),并进行干预,避免 SMA 出生缺陷的发生;二级预防通过产前诊断减少SMA患者的出生;三级预防通过对新生儿/儿童筛查,实现早期发现,早期诊断,早期干预。
孕前携带者筛查是防控核心措施。备孕期夫妻,尤其有相关家族病史的高危人群,应提前开展基因筛查。若夫妻双方均为携带者,每次怀孕都有25%的几率生育SMA患儿,50%的几率生育致病基因携带者,25%的几率生育健康孩子。
高危家庭孕期需及时行产前诊断,通过羊水、绒毛、脐血检测判断胎儿基因状态。同时,胚胎植入前遗传学诊断(PGD)技术,可在胚胎移植前筛选健康胚胎,从源头杜绝SMA患儿出生。
SMA作为罕见遗传病,给患者及其家庭带来沉重的负担。了解SMA的相关知识,重视预防和早期诊断,积极采取有效的治疗措施,对于改善患者生活质量、延长生存期具有重要意义。在此呼吁全社会共同关注,给予SMA患者更多的关注和支持,共同推动罕见病领域的医学进步和发展。